Scientific Challenge leader: Edoardo Di Napoli, Alessandro Pecchia
This project aims to upgrade libNEGF into the exascale domain, creating
the first freely available library tool able to tackle large-scale calculations
of non-equilibrium charge carrier transport. This will be the first tool to
fully encompass all the relevant interaction mechanisms in order to obtain
realistic simulations of nanoscale optoelectronic devices.
The
applications of libNEGF will target 2D heterostructures, traditional III- V
multi quantum-wells, nano-electronic device simulations (both 2D and 3D) as
well as nano-thermoelectric devices.
The goal
is to expand the libNEGF library to create a versatile quantum simulation tool
that can handle industry-relevant simulations of optoelectronic
hetero-structures based on 2D materials with DFT level accuracy. This includes
accounting for light-carrier interactions, electron-phonon scattering, and
excitonic effects. To achieve this, the libNEGF code must be made scalable on
pre-exascale and future exascale systems by developing new algorithms, data
distribution methods, and platform-aware kernels.
The NEGF
formalism is a computationally intensive method dealing in essence with large
dense tensors in configuration, energy, and momentum space. To address the
memory scaling issue, strategies such as data distribution, data pipelining, overlapping
of communication and computation, and reduced-precision storage of intermediate
results must be implemented.
We aim
introducing further layers of parallelization and distribution, develop a
flexible block-dense structure scheme, consider the effect of data compression
and accuracy, and design a new data flow strategy that maximizes the
interleaving of computation with communication. In addition, we aim at
achieving multi-level parallelism and interoperability between different
platforms, such as NVIDIA, Intel, AMD, ARM, etc., by developing an automatic
framework that combines compiler-based code generation with heuristic
performance prediction tools that evaluate the generated code through modeling
of the computation and communication patterns of the underlined algorithm.
LibNEGF is
already interfaced to DFT codes such as dftb+ or the finite-element device
simulator tiberCAD. Additional interfaces are in progress, with BigDFT with a
wavelet or LCAO basis sets, or tools exploiting maximally-localized Wannier
representation of the local basis.
To
validate the tool’s capabilities and ability to perform at exascale, it will be
tested on a few identified structures, such MoS2/WS2
multilayers and MoS2/graphene with varying compositions and
transport orientations. The tests will focus on exciton splitting ability and
internal quantum efficiencies, representing the first demonstration of NEGF
simulations on large-scale hetero-structures of this type.
Metalwalls is a classical molecular dynamics code aiming at simulating electrochemical cells. It treats electrode as metallic systems held at constant potential and includes polarization effects for the liquids.
libNEGF is a general library that calculates Equilibrium and Non Equilibrium Green’s Function and related quantities in open systems, within an efficient sparse iterative scheme. The code will be optimized to describe photon-carrier dynamics (generation, transport and recombination) in nanostructured regions and at complex interfaces, and used to execute advanced simulation of high-efficiency solar cell devices for different input biases.
KMC/DMC (http://people.bath.ac.uk/pysabw/). Kinetic Monte Carlo (KMC) simulates charge and energy transport in organic solar cells. Device Monte Carlo (DMC) simulates charge transport in perovskite cells. The DMC code includes the effects of mobile ion motion that affects most perovskites.
The most important aspect of the Material for Energy scientific challenge of EoCoE is to join scientific and technical competences from the most important institutions and research centres in Europe in order to discover novel materials that allow the production of electricity by harvesting energy from sunlight, salinity gradients and temperature gradients. In order to achieve this challenging purpose, we are developing codes based on theories at different length scales able to be run efficiently on the next generation of supercomputers that are reaching exascale computing performance, i.e., supercomputers able to perform one billion billion (1018) floating-point operations per second.
Thanks to the EoCoE project, four different European Institutions (CIEMAT, CNR, ENEA and FZJ) collaborated in understanding the transport mechanisms underlying photovoltaic devices based on the Silicon Heterojunction technology. Amorphous-crystalline heterointerfaces play a crucial role in the photovoltaic operation of the devices, but the microscopic mechanisms of transport and recombination mechanisms at the interface are not yet well understood. The main purpose here is to design and develop cutting-edge computational methods and HPC-ready software for photovoltaic device simulations at atomistic resolution in the most insightful and predictive way. The results look very promising for further research since we have simulated the transport at the interface between passivated Silicon and amorphous Silicon by taking into account the presence of defects in a realistic structure, and increased the speed of specific algorithms used by the code more than six times. Moreover, EoCoE has signed a MoU with MAX CoE to collaborate on the optimization of the pre-processing of the electronic properties (using QUANTUM ESPRESSO) before inserting the definition of these properties in the transport calculation.
Another objective reached by the collaboration of CEA and Maison de la Simulation is to assess the best electrode/electrolyte combination to optimize the electricity production in electrochemical systems. Electrical power production from salinity gradients harvests the free energy lost during the mixing of sweet water with salty water in river estuaries. The main technologies developed for this purpose to date exploit the electric potential differences applied across membranes. We analyse an alternative approach based on capacitive mixing. As part of this approach, we are investigating the electrode structure which optimizes a blue energy production, and which is the compositions that will enable optimal performances of thermo-electrochemical cells. Five-hundred structures never seen before were passed through the artificial intelligence model to predict the vertical energy gaps between the reduced and the oxidized species, necessary to predict electrochemical properties (e.g. redox potentials). Such a tool has the potential to accelerate the research of the best electrode/electrolyte combination to optimize the energy harvesting process.
Working with the University of Bath (research group of Prof. Alison Walker), we have been able to achieve good results on optoelectronic devices based on two classes of emerging photovoltaic materials, namely organic molecular semiconductors and metal-halide perovskite semiconductors. Both types of cells are cheap to make but have some stability concerns. Organic solar cells are only 12% efficient whereas perovskite solar cells are close to 23% efficiency. However, perovskite cells are notorious for poor stability and current-voltage hysteresis. Our purpose is to design and develop HPC ready software for device models of both cell types extending the simulations to systems of several thousands of atoms to understand degradation mechanisms and find materials and architectures that enhance stability.
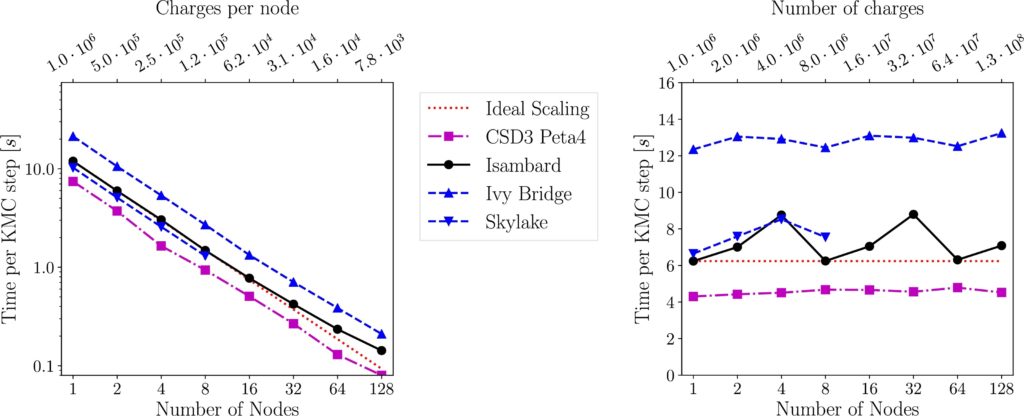
The specificities of these two materials necessitate different computational approaches. Therefore, the development, implementation and application of simulation methodologies amenable to studying phenomena at a broad range of length and time scales has evolved. We are developing a pair of Monte-Carlo codes (KMC and DMC). Notably, algorithmic improvements and other optimization work on the KMC code enabled access to organic molecular semiconductors models that contain millions of charges with good parallel scaling. We demonstrated good performance scaling for 128 million charges up to 128 nodes on Intel and ARM architectures, making possible to model doped organic semi-conductor containing 20,000 charges, while the previous state of the art code, using approximate electrostatics, could perform KMC simulations with systems containing less than 100 charges.
Metal-halide perovskites are currently attracting intense interest for application in optoelectronic devices, due to their remarkable efficiencies in spite of low temperature synthesis methods. In order to test the influence of large polarons on the value of phenomenological parameters, we derived expressions for scattering of polarons by ionised impurities and acoustic phonons, and implemented them in the DMC simulator in order to compute and compare the temperature dependent mobilities of polarons and band electrons in three metal–halide perovskites. We investigated the role played by polaron formation and we found that the main effect is to reduce the mobility of electrons, but the temperature dependence is not altered. This is important for understanding the degradation mechanisms and for the design of solar cells based on metal-halide perovskites with enhanced stability in the performance of the device.
The figures below are snapshots of the results published in scientific articles. Please refer also to the publications section for more details.
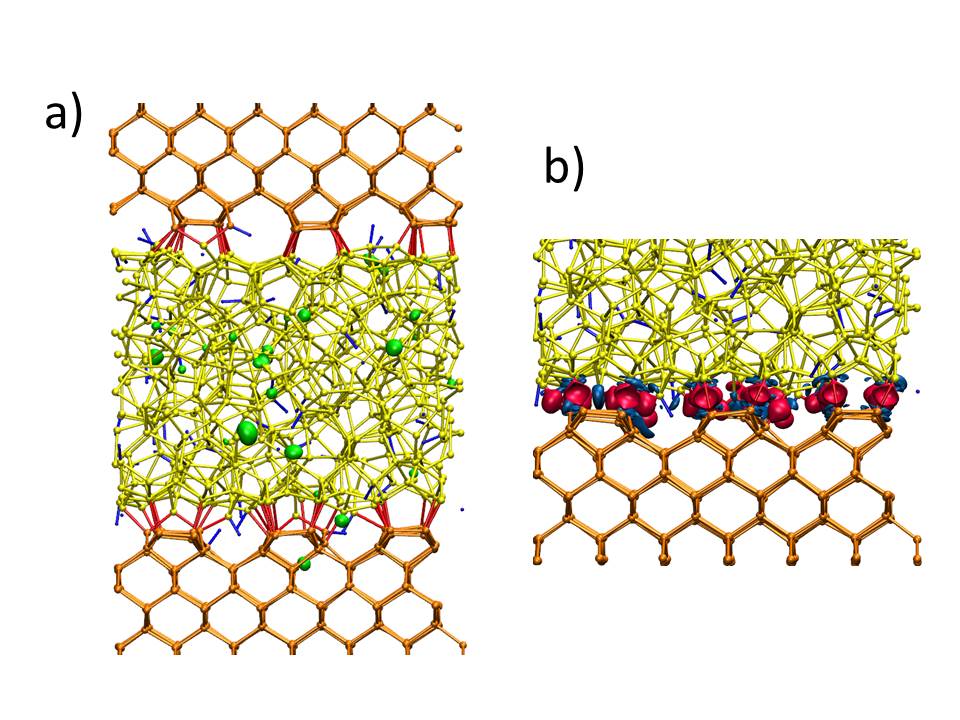
Fig. 1. Towards exascale simulations of a-Si:H/c-Si interfaces. a) Local density of states (green isosurface) of the intragap states of a-Si:H/c-Si interface at room temperature. They correspond to defect states localized both in the bulk of a-Si and at the a-Si:H/c-Si interface. The Silicon atoms and their bonds are in orange in the c-Si side and are in yellow in the a-Si:H side, Hydrogen atoms and bonds with Silicon atoms are in blue. Bonds between c-Si and a-Si are in red. a-Si = amorphous Silicon; c-Si = crystal Silicon. b) The c-Si/a-Si:H interface and the difference at of the charge density between the total system and the c-Si and a-Si:H systems considered as isolated slabs at the end of the room temperature thermalisation. Red (blue) isosurface is positive (negative) difference of the charge density. The electron charge is accumulated along the c-Si/aSi:H interface and depleted from the nearby c-Si and a-Si:H surfaces. (Article from Francesco Buonocore, Massimo Celino, Pablo Luis Garcia, Simone Giusepponi, Rafael Mayo-Garcia. WSC 20: Proceedings of the 2020 Winter Simulation Conference, IEEE press (accepted), http://meetings2.informs.org/wordpress/wsc2020/)
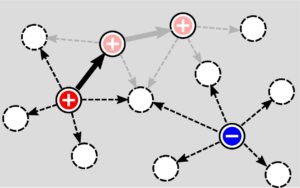
Fig. 2. Organic and perovskite solar cells. Schematic sketch of one KMC step, which consists of calculation of the propensities for all possible hops (dashed arrows) and moving one particle to a new site after accepting a particular hop (solid arrows). Subsequent steps are shown as grey arrows. Kinetic Monte Carlo (KMC) simulations of a system of (electrically) charged particles require many evaluations of the change in electrostatic energy associated with “possible” moves of individual particles, but only one such move is selected per KMC step. (from article by W. R. Saunders, J. Grant, E. H. Muller and I. Thompson, J. Comp. Phys. 410, 109379 (2020) DOI: 10.1016/j.jcp.2020.109379)
- William Robert Saunders, James Grant, Eike Hermann Müller, Ian Thompson, Fast electrostatic solvers for kinetic Monte Carlo simulations, Journal of Computational Physics, Volume 410, 2020, 109379, ISSN 0021-9991, https://doi.org/10.1016/j.jcp.2020.109379.
- James M. Cave, Nicola E. Courtier, Isabelle A. Blakborn, Timothy W. Jones, Dibyajyoti Ghosh, Kenrick F. Anderson, Liangyou Lin, Andrew A. Dijkhoff, Gregory J. Wilson, Krishna Feron, M. Saiful Islam, Jamie M. Foster, Giles Richardson, and Alison B. Walker. Deducing transport properties of mobile vacancies from perovskite solar cell characteristics. Journal of Applied Physics 128, 184501 (2020), https://doi.org/10.1063/5.0021849
- Awad Shalabny, Francesco Buonocore, Massimo Celino, Gil Shalev, Lu Zhang, Weiwei Wu, Peixian Li, Jordi Arbiol, and Muhammad Y. Bashouti. Semiconductivity Transition in Silicon Nanowires by Hole Transport Layer. Nano Letters 20, 11 (2020), 8369–8374, https://doi.org/10.1021/acs.nanolett.0c03543
- Marin-Laflèche et al. MetalWalls: A classical molecular dynamics software dedicated to the simulation of electrochemical systems. Journal of Open Source Software, 5(53), 2373, https://doi.org/10.21105/joss.02373
- Michele Ruggeri, Markus Holzmann, David M. Ceperley, and Carlo Pierleoni, Quantum Monte Carlo determination of the principal Hugoniot of deuterium. Phys. Rev. B 102, 144108 (2020), https://doi.org/10.1103/PhysRevB.102.144108
- Das, B; Aguilera, I; Rau, U; Kirchartz, T. What is a deep defect? Combining Shockley-Read-Hall statistics with multiphonon recombination theory. Physical Review Materials Volume: 4 Issue: 2 Article Number 024602. https://doi.org/1103/PhysRevMaterials.4.024602
- Barberio, S. Giusepponi, S. Vallières, M. Scisció, M. Celino, P. Antici. Ultra-fast High-precision Metallic nanoparticle Synthesis using Laser-Accelerated protons. Scientific Reports (2020) 10:9570. https://doi.org/10.1038/s41598-020-65282-9
- Antonio Riquelme, Laurence J. Bennett, Nicola E. Courtier, Matthew J. Wolf, Lidia Contreras-Bernal, Alison B. Walker, Giles Richardson and Juan A. Anta. Identification of recombination losses and charge collection efficiency in a perovskite solar cell by comparing impedance response to a drift diffusion model. Nanoscale, 2020, 12, 17385–17398. https://doi.org/10.1039/D0NR03058A
- Laurence M.Peter, Alison B.Walker,Thomas Bein, Alexander G. Hufnagel, Ilina Kondofersky. Interpretation of photocurrent transients at semiconductor electrodes: Effects of band-edge unpinning. Journal of Electroanalytical Chemistry, Volume 872, 1 September 2020, 114234. https://doi.org/10.1016/j.jelechem.2020.114234
- Kyle G. Reeves, Alessandra Serva, Guillaume Jeanmairet, Mathieu Salanne. A first-principles investigation of the structural and electrochemical properties of biredox ionic species in acetonitrile. Physical Chemistry Chemical Physics 2020,22, 10561-10568. https://doi.org/10.1039/C9CP06658
- All the EoCoE-II publications are available here (OpenAIRE).
Work in progress