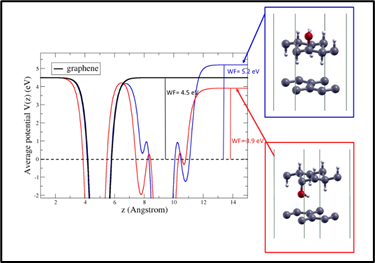
Abstract: The thermodynamic stability of hydroxylated graphane, that is, fully sp3 graphene derivatives coordinated with -H and -OH groups, has been recently demonstrated by ab initio calculations. Within the density functional theory approach, we investigate the electronic property modifications of graphane by progressive hydroxylation, that is, by progressively substituting -H with -OH groups. When 50% of graphane is hydroxylated, the energy bandgap reaches its largest value of 6.68 eV. The electronic affinity of 0.8 eV for graphane can widely change in the 0.28-1.60 eV range depending on the geometric configuration. Hydroxylated graphane has two interfaces with vacuum, hence its electron affinity can be different on each interface with the formation of an intrinsic dipole perpendicular to the monolayer. We envisage the possibility of using hydroxylated graphane allotropes with tunable electronic affinity to serve as interfacial layers in 2D material-based heterojunctions.
Francesco Buonocore, Andrea Capasso, Massimo Celino, Nicola Lisi and Olivia Pulci
J. Phys. Chem. C 2021, 125, 16316−16323